@tgof137: A few people asked me about No...
@tgof137
19 views
Mar 31, 2026
Advertisement
1
A few people asked me about Nod's preprint about 2 spillovers.
He makes two arguments criticizing Pekar et al 2022.
A proper analysis of his 1st argument actually points in the opposite direction and strengthens Pekar's conclusions.
His 2nd argument is not well defined.
🧵
He makes two arguments criticizing Pekar et al 2022.
A proper analysis of his 1st argument actually points in the opposite direction and strengthens Pekar's conclusions.
His 2nd argument is not well defined.
🧵
2
3
The outbreak in Wuhan is unusual.
Normally, when a single case of Covid starts an outbreak, it starts a single polytomy. We've observed this happening again and again, around the world.
In Wuhan, there are 2 polytomies. Pekar theorized that was from 2 spillovers.
Normally, when a single case of Covid starts an outbreak, it starts a single polytomy. We've observed this happening again and again, around the world.
In Wuhan, there are 2 polytomies. Pekar theorized that was from 2 spillovers.

4
Because there are only 2 mutations separating the 2 lineages, this could also happen by chance, from 1 spillover.
Pekar's model said that was a rare thing to occur, with 3% odds from a single introduction.
Pekar's model said that was a rare thing to occur, with 3% odds from a single introduction.
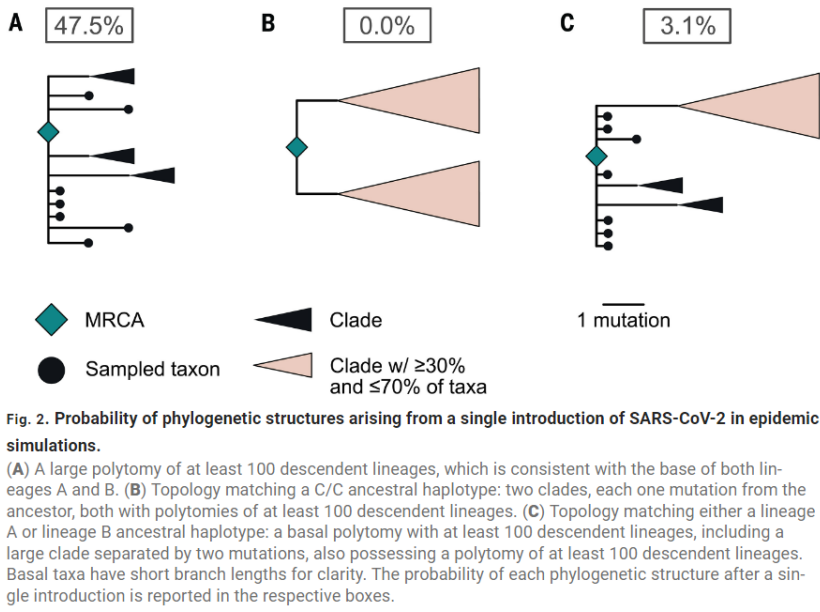
5
(the odds are actually even lower than that, because the genetic clock is reversed. A spilled over after B. So it's probably closer to 0.3%, but let's ignore that for now)
6
How often do you get 2 polytomies from 2 spillovers?
The simplest calculation would be you just simulate 2 epidemics, and count how often both form basal polytomies
That gets you to a bayes factor of somewhere around 4X, in favor of 2 spillovers, not 1:
The simplest calculation would be you just simulate 2 epidemics, and count how often both form basal polytomies
That gets you to a bayes factor of somewhere around 4X, in favor of 2 spillovers, not 1:
View Tweet
7
Nod points out an additional constraint that Pekar missed.
Pekar required that the 2 polytomies both need to be equal and balanced, with each making 30-70% of the total genomes.
Pekar required this for the 1 introduction case but did not require it for the 2 introduction case.
Pekar required that the 2 polytomies both need to be equal and balanced, with each making 30-70% of the total genomes.
Pekar required this for the 1 introduction case but did not require it for the 2 introduction case.
8
If you look at the start time of all possible simulated epidemics, some grow really slowly and some grow quickly.
If you randomly pick 2 points from that curve, the two aren't always going to be close to each other.
If you randomly pick 2 points from that curve, the two aren't always going to be close to each other.
View Tweet
9
At this point, you should probably stop and think about what we're actually modeling here.
We're saying that it is possible for an epidemic that starts some random place in Wuhan to grow very slowly before it takes off.
We're saying that it is possible for an epidemic that starts some random place in Wuhan to grow very slowly before it takes off.
10
Maybe it starts in late September or early October, goes from person to person for a few weeks, and then takes off when it hits Huanan market.
That might represent something like, "one infected person from Yunnan visits Wuhan and starts the pandemic":
That might represent something like, "one infected person from Yunnan visits Wuhan and starts the pandemic":
View Tweet
11
What we're actually trying to model here is "2 spillovers at Huanan market". Everyone agrees the market is a reasonably good place for Covid to spread.
Some lab leakers think the market is the perfect place for Covid to spread, better than any other.
Some lab leakers think the market is the perfect place for Covid to spread, better than any other.
12
That part is debatable, I don't know how much better or worse it is than any other crowded building.
But we all agree it's a crowded building, so an introduction there quickly starts growing.
But we all agree it's a crowded building, so an introduction there quickly starts growing.
13
No one thinks that a spillover at the market is just going to start in September, bounce around from one person to one person for 2+ months, and then suddenly start growing quickly.
14
Pekar's code simulates transmission across a social network. Some nodes in that network are well connected, others are poorly connected. The slowly growing epidemics start at a poorly connected node and jump along such nodes for a while before hitting a well connected node.
15
If you simulate 2 spillovers at the Huanan market as two introductions into random places in that social network, then it's not a model that represents reality.
16
I ran some numbers here, with my own code. Suppose I simulate 2 spillovers, with lineage A and lineage B, at the same time.
Suppose those are completely independent of each other, i.e. they happen in different locations.
Suppose those are completely independent of each other, i.e. they happen in different locations.
17
I counted only the cases where both spillovers did not go extinct.
I found it's about 9% odds that the two will become balanced at 30/70 or closer.
If I just divide 9% by 3%, I get bayes factor 3.
So, by that logic, Nod did find something, he reduced ~4 down to ~3.
I found it's about 9% odds that the two will become balanced at 30/70 or closer.
If I just divide 9% by 3%, I get bayes factor 3.
So, by that logic, Nod did find something, he reduced ~4 down to ~3.
18
But, if we suppose a market outbreak is faster than a random introduction somewhere in Wuhan, then we actually want to put some constraint on that.
How should we quantify that?
How should we quantify that?
19
One approach would be to say a market spillover has no mutations before the basal polytomy forms.
Those epidemics happen a little faster than average, but they can still take a few generations to get going. Here's an example, with TMRCA marked by the orange line.
Those epidemics happen a little faster than average, but they can still take a few generations to get going. Here's an example, with TMRCA marked by the orange line.

20
Another approach would be to count epidemics where the TMRCA happens right after the first case.
These aren't so uncommon in general (I think it's ~33% of all simulations), and might be a good representation of what a introduction looks like in a high spread environment.
These aren't so uncommon in general (I think it's ~33% of all simulations), and might be a good representation of what a introduction looks like in a high spread environment.

21
If we represent a market spillover the first way, as an epidemic with a basal polytomy, then it's 36% likely that you get the two balanced polytomies.
36/3 = 12, so the bayes factor is 12X in favor of 2 spillovers instead of 1.
36/3 = 12, so the bayes factor is 12X in favor of 2 spillovers instead of 1.
22
If you represent a market spillover the second way, the odds favoring 2 spillovers would be even higher still. (I did not measure this yet in my code)
23
Either way, I think you're going to end up with 10X or higher odds that "2 market spillovers produce 2 balanced polytomies", as opposed to "1 introduction somewhere in Wuhan produces 2 balanced polytomies"
24
That's interesting, I might not have thought about it if Nod hadn't brought it up.
But I'm glad he started this discussion and helped me understand that Pekar actually underestimated how likely 2 spillovers is!
But I'm glad he started this discussion and helped me understand that Pekar actually underestimated how likely 2 spillovers is!
25
The problem here is that the lab leak theory has conflicting goals.
Most of the people supporting the lab leak theory want to say that the market outbreak is meaningless, because markets are just really good at spreading Covid.
Most of the people supporting the lab leak theory want to say that the market outbreak is meaningless, because markets are just really good at spreading Covid.
26
Rootclaim actually argued that the Huanan market was the single most likely place in all of Wuhan for a lab leak to be discovered.
While simple math says the lab leak showing up at the raccoon dog shop is a 1 in 10,000 coincidence, they gave it 100% odds of happening.
While simple math says the lab leak showing up at the raccoon dog shop is a 1 in 10,000 coincidence, they gave it 100% odds of happening.
27
Meanwhile, Nod is trying to make an argument that maybe some market outbreaks grow really slowly and some grow really quickly, therefore 2 market spillovers will not end up with the same size.
28
If you support the lab leak theory, you can not simultaneously believe both of these things.
You can make an argument that the market outbreak happening after a lab leak is higher, but if you do that, you can't also make Nod's argument.
You can make an argument that the market outbreak happening after a lab leak is higher, but if you do that, you can't also make Nod's argument.
29
This is a really common problem, for the lab leak theory. Elsewhere, they try to argue that the leak happened on September 12th (because some "database went down") but that it also happened in mid November (because "3 people got sick at the Wuhan lab")
30
In reality the lab leak theory is just a long series of observations that sound suspicious on their own, but none of them fit together when you try to combine them.
There's no one cohesive lab leak theory that makes sense.
There's no one cohesive lab leak theory that makes sense.
31
There's a second half of Nod's argument which isn't as simple to analyze, I'll get into that later.
32
The second half of Nod's argument basically complains that Pekar gave the animals "viral diversity for free".
For the one introduction case, the virus needs to have 2 early mutations, and then both clades grow quickly to become balanced.
That's rare, only 3% likely.
For the one introduction case, the virus needs to have 2 early mutations, and then both clades grow quickly to become balanced.
That's rare, only 3% likely.
33
For the 2 spillovers case, the animals already have a certain amount of diversity, and you're just selecting 2 spillovers from that pool of diversity.
This diagram from Pekar et al 2022 shows roughly how it works, and also shows how the "clock reversal" can happen.
This diagram from Pekar et al 2022 shows roughly how it works, and also shows how the "clock reversal" can happen.

34
One problem here is that Pekar never modeled that diversity among the animals, and just assumed that 2 mutations between the 2 spillovers is a reasonable amount to happen.
But... should you actually model that diversity, and take 2 random samples from it?
But... should you actually model that diversity, and take 2 random samples from it?
35
Like, if it was as seen in that diagram that Pekar drew, the 2 spillovers could have 2 mutations apart. But they could also have 0, or 1, or more than 2.
36
If you pick 2 spillovers at random, do you need to weigh the relative odds of 2 vs 1 vs 0 mutations?
In that case, Nod would say that Pekar should reduce the odds, because 2 mutations is possible, but it might be too many.
In that case, Nod would say that Pekar should reduce the odds, because 2 mutations is possible, but it might be too many.
37
There's been a similar complaint by lab leak supporters in the past.
In this comment, Virginie says that 2 mutations is too few, and she thinks that 5 mutations would be more reasonable:
In this comment, Virginie says that 2 mutations is too few, and she thinks that 5 mutations would be more reasonable:

38
But, notice that is the opposite complaint!
It's hard to take this seriously when Nod says that 2 mutations is too many and the other lab leakers say that 2 mutations is not enough.
It's hard to take this seriously when Nod says that 2 mutations is too many and the other lab leakers say that 2 mutations is not enough.
39
Anyways, maybe you could try to simulate it, and consider various amounts of diversity in the animals.
I think that's what Nod is trying to do.
His writing is not particularly clear and I haven't read his code.
I think maybe what he's trying to do here is:
I think that's what Nod is trying to do.
His writing is not particularly clear and I haven't read his code.
I think maybe what he's trying to do here is:
40
1. "assume that the market animals start with one infected animal"
2. "simulate the growth and diversity in the number of animal cases as if that was a human epidemic"
3. "pick 2 animals at random for spillover"
2. "simulate the growth and diversity in the number of animal cases as if that was a human epidemic"
3. "pick 2 animals at random for spillover"
41
Is that approach valid?
I don't know. Did the market outbreak start with a single infected animal or many?
Do animal outbreaks grow via the same model as human outbreaks?
Pekar is simulating along a network of human social connections. Do raccoon dogs have the same network?
I don't know. Did the market outbreak start with a single infected animal or many?
Do animal outbreaks grow via the same model as human outbreaks?
Pekar is simulating along a network of human social connections. Do raccoon dogs have the same network?
42
One way to test this might be to look at the Hong Kong example that Virginie mentioned.
In that case, there were 2 spillovers from 2 Covid infected hamsters to people. The 2 spillovers had 5 mutations apart.
In that case, there were 2 spillovers from 2 Covid infected hamsters to people. The 2 spillovers had 5 mutations apart.
43
Let's assume that the hamster shop started with a single infected hamster, and the viral diversity grew from there, with exponential spread across the hamster social network.
On average, it takes about 60 days to get 5 mutations.
On average, it takes about 60 days to get 5 mutations.
44
In 60 days of spread, 1 human infection turns into about 60,000 infections.
We can thus conclude that there were 60,000 infected hamsters in that shop.
We can thus conclude that there were 60,000 infected hamsters in that shop.
45
Of course, that's not the case at all, there were far fewer animals than that.
They gained viral diversity in prior transmission in warehouses, etc. They were mixed up, moved around, and eventually put in one shop. There were 2 shipments from the Netherlands to Hong Kong.
They gained viral diversity in prior transmission in warehouses, etc. They were mixed up, moved around, and eventually put in one shop. There were 2 shipments from the Netherlands to Hong Kong.
46
So, what actually happened at Huanan market? 1 infected animal came in? A group of animals with some diversity came in? 2 different shipments came in, one week apart?
47
I'm not sure that any of this is spelled out well enough to put clear odds on it. But I haven't thought about Nod's 2nd argument for long enough to have a strong opinion.
48
So, that's Nod's 2 new arguments. The first is wrong and the second is very poorly defined.
I expect that Nod will come back with more explanations for why these are actually genius ideas. And maybe I'm missing something here. But so far I'm not seeing much.
I expect that Nod will come back with more explanations for why these are actually genius ideas. And maybe I'm missing something here. But so far I'm not seeing much.
49
I think there's this fantasy that if they just pull one thread on the market hypothesis, the whole thing unravels and lab leak becomes true.
But it's mostly just increasingly obscure arguments to try to explain away the obvious bullseye on Huanan market.
But it's mostly just increasingly obscure arguments to try to explain away the obvious bullseye on Huanan market.
View Tweet
50
If you want to argue that Covid started somewhere other than that market, you should actually try to answer questions like:
"when did it start?"
"where did it start?"
"which MRCA?"
"how much time/how many cases before A/B split?"
And then compare that theory to real Wuhan data.
"when did it start?"
"where did it start?"
"which MRCA?"
"how much time/how many cases before A/B split?"
And then compare that theory to real Wuhan data.
51
I would encourage Nod to answer those questions.
52
I tried running thousands of simulations until I got a few clock reversals from 1 introduction. Here's what the most common phylogeny looks like.
The 2 mutations happen in a single step, very quickly after Covid starts, then lineage B (blue) outgrows A (red) for random reasons.
The 2 mutations happen in a single step, very quickly after Covid starts, then lineage B (blue) outgrows A (red) for random reasons.


53
Though sometimes it's a little bit more complex, like there are a few more cases before the split happens.

54
So this is great, already, because Nod's argument is that it's "unlikely for 2 mutations to happen in animals".
But instead he's just arguing that the 2 mutations happened immediately after spillover, by some unlikely random chance.
But instead he's just arguing that the 2 mutations happened immediately after spillover, by some unlikely random chance.
55
The median number of cases before the A/B split happens was 16. The median time from introduction to the split was 13 days.
(Those numbers are not perfect, because I only had a few of these reversed clock epidemics to compute from -- these are extremely rare)
(Those numbers are not perfect, because I only had a few of these reversed clock epidemics to compute from -- these are extremely rare)
56
I had to run thousands of simulations to find a few like this, because these are so rare. Pekar said 3% of single introductions form the 2 polytomies by chance, but the odds of also getting the clock reversal are even lower:
View Tweet
57
So your lab leak scenario is basically... A and B split after ~16 cases, and B immediately gains 2 mutations. Then B goes to the market within another ~10-20 cases.
Is it unlikely that B would go to Huanan market, instead of any other place in Wuhan without suspicious animals?
Is it unlikely that B would go to Huanan market, instead of any other place in Wuhan without suspicious animals?
58
I mean... Nod's argument assumes every person in Wuhan is equivalent and a market introduction is identical to any place in Wuhan, so I get to argue for 1 in 10,000 odds.
(~1,000 vendors at Huanan out of 10 million people in Wuhan)
(You can bump that down, but then Nod is wrong)
(~1,000 vendors at Huanan out of 10 million people in Wuhan)
(You can bump that down, but then Nod is wrong)
59
Next up, lineage A gets found on December 15th, in someone living very near the market, at a time when there are probably only ~200 infected people in Wuhan:
View Tweet
60
And then you've got the lineage A sample at the market itself.
Koopmans' map shows that a vendor at that shop had Covid on December 15th.
Koopmans' map shows that a vendor at that shop had Covid on December 15th.
View Tweet
61
If that vendor had lineage A on December 15th, then the argument is just basically over, covid started at the market.
But maybe you can still keep other theories, by saying that market sample was deposited later.
But maybe you can still keep other theories, by saying that market sample was deposited later.
62
The actual odds of the scenario that the lab leak people prefer are even worse than this, because they also think that there's some lineage that came before A, and they think that there are intermediate genomes.
I drew in what this would hypothetically look like:
I drew in what this would hypothetically look like:

63
So now you're not just arguing for a rare "clock reversal" but an even rarer "double clock reversal", where A outpaces A+29095T (by chance), then B outpaces both (by chance).
And somewhere along the way, there were intermediates (even though none of the simulations had those)
And somewhere along the way, there were intermediates (even though none of the simulations had those)
64
When you get 2 polytomies from 1 introduction, that can happen either from a single case having 2 mutations, or from 2 cases having 1 introduction and then the intermediate clade dies out.
It's usually the first case, not the latter.
It's usually the first case, not the latter.
65
And the one thing that basically never happens is that the intermediate clade exists but it grows so slowly that it's only discovered months later, in February.
If it does exist, it either dies out or grows to become very large:
If it does exist, it either dies out or grows to become very large:
View Tweet
66
The simulations where it's something in between are < 1%.
So now the lab leak scenario is like:
3% for 2 polytomies *
< 10% for one clock reversal *
< 10% for second clock reversal *
< 1% for T/T intermediates that exist but don't grow
So now the lab leak scenario is like:
3% for 2 polytomies *
< 10% for one clock reversal *
< 10% for second clock reversal *
< 1% for T/T intermediates that exist but don't grow
67
And that's just to get the weird phylogeny that lab leakers imagined happened from 1 introduction, not even counting the odds that B went to the market and A probably did too.
68
So, Weissman is going to come in now and say "blah blah blah, use ratios not odds"
And he will be correct, I need to model both and calculate the ratios for all these things.
And he will be correct, I need to model both and calculate the ratios for all these things.
69
But most of these things will obviously go in my favor -- the phylogeny makes sense if B came before A, which came before A+29095T, rather than the lab leak world where everything is backwards and requires rare coincidences at every step.
70
So, yeah, it is worth doing more calculations and making this as rigorous as possible.
But I'm pretty confident that any reasonable model will still put the 2 spillovers hypothesis way ahead, and the A0 -> A -> B0 -> B scenario described in Lv et al 2024 will come out behind.
But I'm pretty confident that any reasonable model will still put the 2 spillovers hypothesis way ahead, and the A0 -> A -> B0 -> B scenario described in Lv et al 2024 will come out behind.